Patrice Koehl
Department of Computer Science
Genome Center
Room 4319, Genome Center, GBSF
451 East Health Sciences Drive
University of California
Davis, CA 95616
Phone: (530) 754 5121
koehl@cs.ucdavis.edu
|
|
| Patrice Koehl |
Computational Structural Biology: Understanding Biomolecular Structure, Function, and Dynamics |
Main collaborators: Marc Delarue (Institut Pasteur, France), Henri Orland (CEA, Paris, France), Sebastian Doniach (Stanford University)
The molecular basis of life rests on the activities of biological macromolecules, mostly nucleic acids and proteins. A perhaps surprising finding that crystallized over the last handful of decades is that geometric reasoning plays a major role in our attempt to understand these activities. Biomolecules have shapes that control their functions; these shapes are defined by a complex network of physical interactions, both internal to the molecule and external through interactions with their aqueous environment. My research addresses this connection between physics, geometry and biology. I am interested in how the morphology of a biological shape is specified and changes over time. I focus on proteins and RNA. I am particularly interested in morphogenesis, i.e. can we predict how molecular shapes form from fundamental physical principles, and in morphodynamics, i.e. in how molecular shapes evolve with time (for example how do we simulate the structural transition between two known forms of a macromolecule?). I give below two examples of my efforts in this field.
Importance of electrostaticsChemistry can essentially be described as the science of interactions between electrons and their associated wave functions. As such, electrostatics is at the core of all chemical and subsequently biological processes. Quantifying electrostatic interactions has been the focus of many research efforts in biophysical sciences for at least one century; yet we are still lacking a set of accurate tools for computing the electrostatic properties of biomolecules immersed in a solvent and surrounded with an ion atmosphere that can be used for the analysis of experiments, for the prediction of the stability, dynamics and function of biomolecules. Such tools are a prerequisite for the design of molecular partners or inhibitors, as in drug design. They need to be solidly anchored into the physical principles that govern biomolecular electrostatics and at the same time be accessible to the biomedical community at large, with minimal practical requirements in term of computing needs. Computing the solvation energy of a biomoleculeSoluble biomolecules adopt their stable conformation in water, and are unfolded in the gas phase. It is therefore essential to account for water in any modeling experiment. Molecular dynamics simulation that include a large number of solvent molecules are the state of the art in this field, but they are inefficient as most of the computing time is spent on updating the position of the water molecule. It should further be noted that it is not always possible to account for the interaction with the solvent explicitly. For example, energy minimization of a system including both a protein and water molecules does not account for the entropy of water, which would behave like ice with respect to the protein. An alternative approach takes the effect of the solvent implicitly into account. In such an implicit solvent model, the effects of water is included in an effective solvation potential, W = Welec + Wnp, in which the first term accounts for the molecule-solvent electrostatics polarization, and the second for the molecule-solvent van der Waals interactions and for the formation of a cavity in the solvent. I have worked with Prof. Edelsbrunner on computing the second term involved in the implicit solvent models described above (see Shapes section). In parallel, I have recently developed in collaboration with Dr Marc Delarue, Institut Pasteur, France, and Dr Henri Orland, CEA, Saclay, an extension to the Poisson Boltzmann formalism, namely the Dipolar Poisson-Boltzmann Langevin (DPBL) model that circumvents the limits of the former as it allows for non uniform dielectric property of the solvent and accounts for the sizes of ions. We have extended this formalism to account for vdW interactions between the water dipoles and we have developed a fast and accurate numerical solver for the corresponding non linear partial differential equations, AquaSol . |
|
|
|
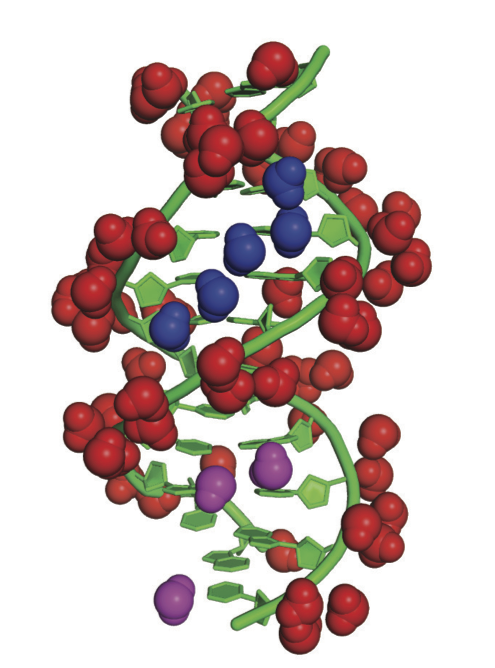
DNA solvation.The Drew-Dickerson dodecamer adopts a right handed double helix structure (green). Its electrostatic potential was computed as the solution of the Dipolar Poisson Boltzmann Langevin equation. The top 72 water molecules corresponding to the highest water density points are represented as solid balls. The positions of these water molecules match remarkably well with the experimental observations. |
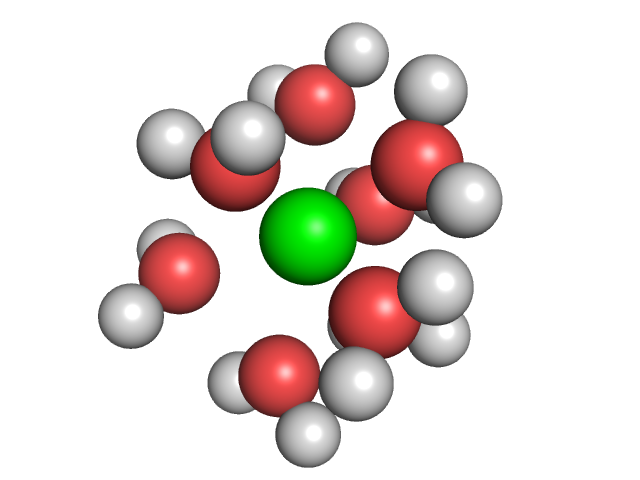
Ion solvation.We computed the electrostatic potential around an isolated magnesium ion using the same Dipolar Poisson Boltzmann Langevin equation. From this potential, we computed the estimated water density around the ion and identified seven regions with high density in the direct neighborhood of the ion. The corresponding seven water molecules match well with the experimental observations on magnesium solvation. |
Related papers:
Structural transitions betwwen two states of a biomoleculeThe functions of many bio-molecules strongly correlate with conformational changes in their structure space, a process usually referred to as their activations. This process for example is very much at the core of enzymatic activity, as an enzyme and its substrate usually go through structural transitions that favor the chemical reaction. The structures of these transition states are of great interest, especially for drug design. Many enzyme inhibitors have been engineered to be transition state analogs, i.e. to resemble the transition state of the enzyme substrate; this design is only possible if the transition state of the enzyme itself is known. This transition state however is very short lived and its structure cannot be studied by standard experimental methods from structural biology. Computational morphing is then a valuable alternative. Classical ``morphing" techniques are linear interpolations of either the Cartesian or the internal coordinates between the initial and end states, followed by energy minimization. With collaborators from Stanford University (Prof. Seb Doniach, Applied Physics) and Institut Pasteur, Paris, France (Dr. Marc Delarue), I have developed a new method, MinActionPath, to calculate the most probable trajectory that is exact for harmonic potentials. This method was illustrated using the classical Elastic Network Model (ENM) to describe both the initial and the final states of the system. The Langevin equation under this potential is solved analytically using the Onsager and Machlup action minimization formalism on each side of the transition, thus replacing the original non-linear problem by a pair of linear differential equations joined by a non-linear boundary matching condition. The crossover between the two multidimensional energy curves around each state is found numerically using an iterative approach, producing the most probable trajectory and fully characterizing the transition state and its energy. |
|
|
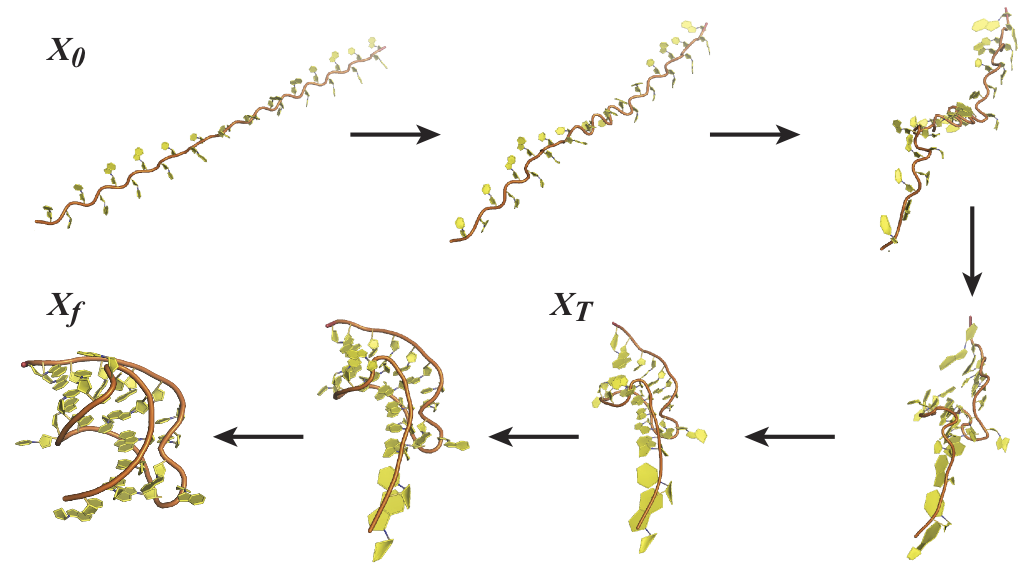
RNA folding:Folding of a small RNA pseudoknot with the path sampling technique MinActionPath under a geometric potential. X0 and Xf are the start and final configurations, respectively, while XT is the predicted transition state. |
Related paper:
J. Franklin, P. Koehl, S. Doniach, and M. Delarue. Minactionpath: maximum likelihood trajectory for large-scale structural transitions in a coarse grained locally harmonic energy landscape. Nucl. Acids. Res., 35:V477-W482, 2007.
| Page last modified 18 July 2017 | http://www.cs.ucdavis.edu/~koehl/ |