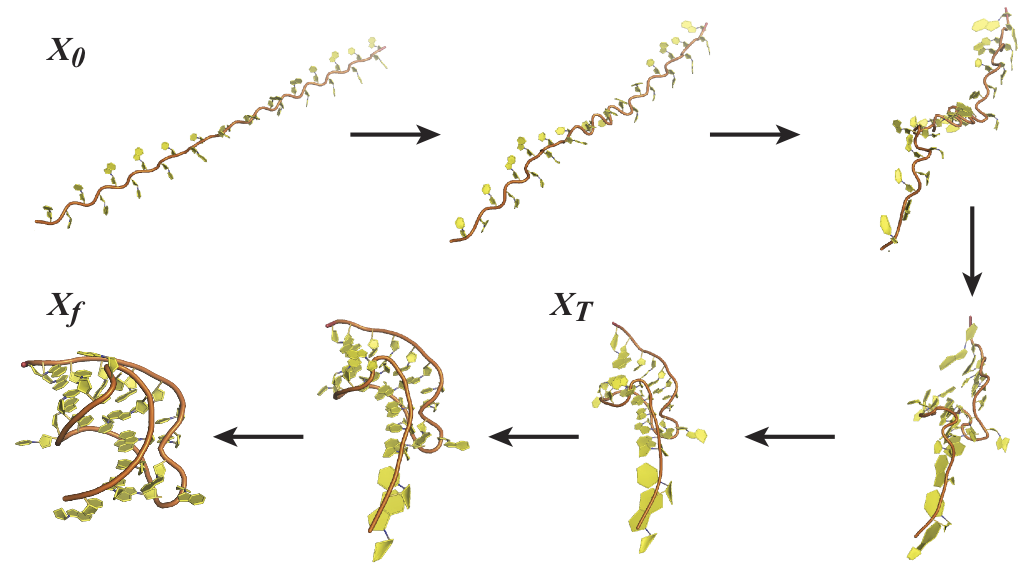
Structural transitions betwwen two states of a biomolecule
The functions of many bio-molecules strongly correlate with conformational changes in their
structure space, a process usually referred to as their activations. This process for example
is very much at the core of enzymatic activity, as an enzyme and its substrate usually go through
structural transitions that favor the chemical reaction. The structures of these transition states
are of great interest, especially for drug design. Many enzyme inhibitors have been engineered
to be transition state analogs, i.e. to resemble the transition state of the enzyme substrate;
this design is only possible if the transition state of the enzyme itself is known.
This transition state however is very short lived and its structure cannot be studied by
standard experimental methods from structural biology. Computational morphing is then a
valuable alternative. Classical ``morphing" techniques are linear interpolations of either the
Cartesian or the internal coordinates between the initial and end states, followed by energy minimization.
With collaborators from Stanford University (Prof. Seb Doniach, Applied Physics) and Institut Pasteur,
Paris, France (Dr. Marc Delarue), I have developed a new method, MinActionPath, to calculate the
most probable trajectory that is exact for harmonic potentials. This method was illustrated
using the classical Elastic Network Model (ENM) to describe both the initial and the final states
of the system. The Langevin equation under this potential is solved analytically using the
Onsager and Machlup action minimization formalism on each side of the transition,
thus replacing the original non-linear problem by a pair of linear differential equations
joined by a non-linear boundary matching condition. The crossover between the
two multidimensional energy curves around each state is found numerically using an
iterative approach, producing the most probable trajectory and fully characterizing the
transition state and its energy.
|